


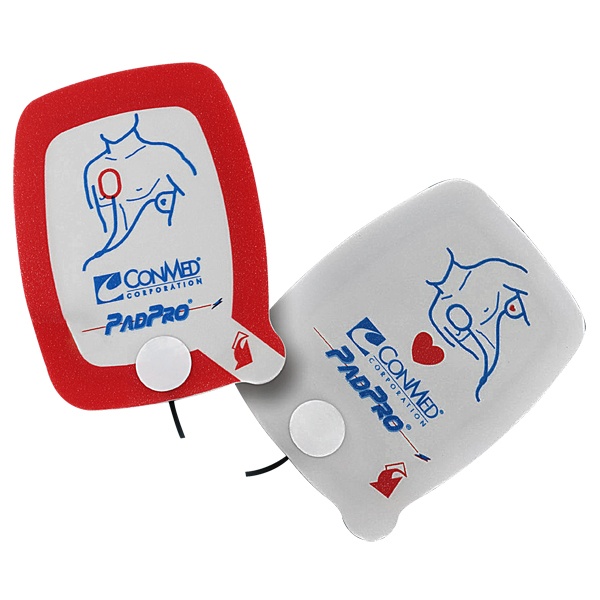
ConMed’s PadPro and R-2 Multi-Function Defibrillation Recalls
ConMed is a medical group with a focus on surgical devices with non-intrusive equipment. The PadPro and R-2 Multifunction Defibrillation devices use electrodes for defibrillation, or correcting heart beats with electric shock, pacing, and monitoring heart activity. ConMed has recently announced a voluntary recall on select devices due to connector compatibility issues. The FDA has declared this to be a Class I recall, which is the most serious recall as it can cause injury or death.
Below is the FDA Firm Press Release:
CONMED Corporation Issues a Voluntary Device Correction for PadPro and R2 Multi-function Defibrillation Electrodes
Contact:
Consumer:
727-399-5276
mutlifunctionelectrodes@conmed.com
FOR IMMEDIATE RELEASE – November 25, 2014 – Utica, New York – CONMED Corporation announces that it has notified customers of a Voluntary Urgent Device Correction for certain PadPro and R2 Multi-function Defibrillation Electrodes due to a connector compatibility issue with Philips FR3 and FRx Defibrillator Units.
These electrodes will not connect with Philips FR3 or FRx AED units. The FRx AED unit requires the pads to be pre-connected, and will issue a continuous alarm chirp to alert the user that the proper pads are not connected to the unit prior to use. The FR3, however, does not require pre-connection and the user will not discover the incompatibility issue until the AED must be used. This may result in a delay in therapy.
CONMED has not received any complaints or Medwatch reports of injuries associated with this Voluntary Device Correction.
The affected multi-function electrodes were distributed globally to distributors and medical facilities from March 1, 2012 through October 29, 2014.
CONMED alerted customers to this issue by letter on November 6, 2014 and is in the process of revising its labeling to clarify the use of these electrodes as incompatible with the Philips FR3 and FRx AED units. There are a total of 174,610 electrodes affected by this voluntary device correction. No product needs to be returned to CONMED.
All lot numbers of the following CONMED electrodes are affected:
Catalog Number 2001H – Adult Radiotransparent Electrode
Catalog Number 2001H-C – Adult Radiotransparent Electrode
Catalog Number 2001H-PC – Adult Radiotransparent Electrode
Catalog Number 2516H – Adult Radiotranslucent Electrode
Catalog Number 2516H-PC – Adult Radiotransparent Electrode
Catalog Number 2603H – Pediatric Radiotranslucent Electrode
Catalog Number 2602H – Mini Pediatric Radiotranslucent Electrode
Catalog Number 3115-1750 – Pediatric R2 Multifunction Electrode
Catalog Number 3115-1751 – R2 Multifunction Electrode
The Food and Drug Administration has classified this as a Class 1 Recall, the most serious recall where there is a reasonable risk of serious adverse health consequences or death.
For further information or to report a problem, please contact CONMED at 727-399-5276, Monday – Friday, 8 am – 5 pm ET, or email at mutlifunctionelectrodes@conmed.com
Health care professionals and customers may report adverse events or quality problems experienced with the use of this product to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail, fax or by phone.
Online: www.fda.gov/medwatch/report.htm
Regular Mail: use postage-paid FDA form 3500 available at: www.fda.gov/MedWatch/getforms.htm. Mail to MedWatch 5600 Fishers Lane, Rockville, MD 20852-9787
Fax: 1-800-332-0178
Phone: 1-800-332-1088
About CONMED
CONMED is a medical technology company with an emphasis on surgical devices and equipment for minimally invasive procedures. Headquartered in Utica, New York, the Company’s 3,600 employees distribute its products worldwide with a direct selling presence in 16 countries outside the United States. International sales constitute approximately 50% of the Company’s revenues.
If you or a loved one have been affected by one of ConMed’s recalled devices, please call us at 1-800-260-2577 for a free consultation.
Source: fda.gov

lawyers are experienced in handling car accident cases and will ensure you get your accident injury claim handeld by an experienced injury lawyer.We can provide you with auto accident attorneys in many cities across the United States of America such as Riverside, Orange County, Los Angeles, San Fernando Valley, Pomona, Ontario, Newport Beach, and San Jose. See our locations section for all cited represented: AA-Accident Attorneys Injury Lawyer Locations. A California Car Accident Lawyer that will fight for you!
